
LA TROMBOCITEMIA ESSENZIALE.
A. Andriani
Nel Lazio si sono formati diversi gruppi di studio che comprendono un gran numero di patologie ematologiche e che si spera siano l’anticamera alla formazione di una vera rete laziale di ematologia che possa ottimizzare le risorse sia umane che economiche.
La formazione di tali gruppi di studio ha permesso di amalgamare tra di loro le diverse ematologie e, nello stesso tempo, di uniformare gli approcci diagnostici e terapeutici. Si stanno, inoltre, unificando le diverse casistiche dei centri che nel tempo porterà sicuramente ad una più accurata valutazione epidemiologica dei diversi gruppi di patologia.
Tra i diversi gruppi di lavoro formatisi vi è quello per si occupa dello studio delle Sindromi mieloproliferative croniche, Ph negative che ha raccolto importanti casistiche di pazienti con trombocitemia essenziale (TE), policitemia vera (PV) e mielofibrosi idiopatica (MF) e che ha permesso al gruppo di presentarsi a convegni nazionali ed internazionali con presentazione di lavori. E’ per questo motivo che si è pensato di fare un aggiornamento su tali patologie che negli ultimi anni hanno presentato notevoli progressi sia in termini biologici che terapeutici.
La TE è caratterizzata dalla produzione midollare di un numero elevato di piastrine e che si manifesta a livello clinico con episodi di trombosi e/o emorragie. L’incidenza di tale patologia è dello 0,59 – 2,53% per 100.000 individui ed i criteri diagnostici della Word Health Organization (WHO) sono riportati in tabella 1; da notare come la classificazione elaborata nel 2008 rispetto a quella del 2001 il numero di piastrine per la diagnosi è stato ridotto a 450.000/mm3 ed è stato inserito dal 2013, anno della scoperta della mutazione di CALR1, 2, 3, anche tale parametro insieme al JAK-2.
|
|
Un dato, che sembra oggi avere importanza da un punto di vista prognostico, è la diagnostica differenziale tra la TE e le fasi precoci della MF4, 5, 6; la diagnostica differenziale si basa su parametri istologici che un patologo abituato a vedere tali patologie non ha difficoltà a riconoscere. Nel futuro sapremo se tale differenziazione ha realmente una importanza clinica: al momento sembrerebbe importante come fattore prognostico sulla sopravvivenza, anche se non sul rischio di sviluppare una trombosi5. Oltre al quadro istologico, che oggi ancora impone l’esecuzione diagnostica della biopsia osteo-midollare all’esordio della malattia, esistono altri parametri biologici che permettono di diagnosticare in modo corretto una sindrome mieloproliferativa cronica; tali esami sono quelli di biologia molecolare quali la determinazione di alcune mutazioni che sono coinvolte nella crescita, maturazione, differenziazione e morte delle cellule staminali midollari: JAK-2, CALR, MPN. La positività di tali mutazioni è presente in circa il 90% delle TE e solo il 10% di pazienti è cosiddetto “triplo negativo”, ossia non esprime nessuna delle mutazioni conosciute e non siamo ancora in grado di definirne la mutazione pato-genetica clonale. Inoltre la presenza di una o dell’altra mutazione conferiscono ai pazienti anche un diverso quadro clinico con differente prognosi3: ad esempio i pazienti con positività del CALR hanno in genere una prognosi migliore e sono caratterizzati da pazienti di sesso maschile più giovani con un più alto numero di piastrine, bassi valori di Hb e globuli bianchi.
Da un punto di vista clinico tale patologia è nella maggior parte dei casi completamente asintomatica alla diagnosi e la scoperta è legata ad esami laboratoristici eseguiti per controllo. In una minoranza di pazienti
l’esordio può essere dovuto alla comparsa di un evento trombotico venoso od arterioso, alla comparsa di fenomeni di tipo Raynaud, disturbi del visus ecc. L’esame obiettivo poco aiuta nella diagnosi se non la presenza, in circa il 20% dei pazienti, di una modesta splenomegalia che in genere è al di sotto dei 5 cm dall’arcata costale7.
La diagnosi di TE è preceduta in circa il 20-25% dei casi da un episodio trombotico che, sicuramente, può incidere sull’approccio terapeutico e sulla prognosi dei pazienti7.
I parametri che si sono riconosciuti importanti da un punto di vista prognostico sono principalmente l’età del paziente alla diagnosi e la presenza o meno di precedenti eventi trombotici. Questi sono gli unici parametri, oltre ad una piastrinosi importante (oltre 1 - 1.500.000 di PLTs/mm3), che inducono il clinico all’inizio di un trattamento specifico. Altri parametri come la positività del JAK-28, il numero dei globuli bianchi9, 10, 11 o la splenomegalia12 sono stati individuati come fattori prognostici per la “Thrombosis Free Survival” (TFS) o l’ “Overall Survival” (OS) ma non sono ancora entrati nella comune pratica clinica.
Le difficoltà alla decisione dell’inizio di un trattamento di tale patologia è legata alla indisponibilità attualmente di farmaci ideali per la cura. La TE ha una prognosi generalmente buona con una sopravvivenza simile a quella della popolazione normale di pari età, almeno nei primi 10 anni dalla diagnosi se ben trattata, per cui bisogna essere molto cauti nel prendere decisioni. Gli episodi di trombosi si verificano in circa il 10% dei pazienti e sono la principale causa di morbidità e mortalità; altre possibili complicanze sono l’evoluzione in MF, la mielodisplasia secondaria/Leucemia acuta e la comparsa di seconde neoplasie primitive.
Le terapie attualmente disponibili sono l’impiego di chemioterapici, l’interferone (IFNα) e l’anagrelide (ANA). Per quanto riguarda il primo gruppo il farmaco che viene abitualmente impiegato è l’idrossiurea (HU), molecola molto ben tollerata della quale a tutt’oggi non si è riusciti a dimostrare una possibile leucemogeneità o l’induzione di un aumentato rischio di sviluppare seconde neoplasie. Al contrario, altri alchilanti quali il piprobromano, o il melphalan avrebbero un aumentato rischio di tali problematiche e per tale motivo andrebbero riservati a pazienti molto ben selezionati, come ad esempio pazienti anziani per i quali si è dimostrata una intolleranza o tossicità da HU.
Oltre alla HU si ha la disponibilità dell’IFNα. Questa molecola, della quale non si conosce ancora bene il meccanismo d’azione (antiproliferativo, immunostimolante, etc.), ha il vantaggio che non essendo un chemioterapico è la molecola di più sicuro utilizzo per i pazienti giovani e per le pazienti che necessitino di un trattamento in gravidanza. E’ l’unica molecola disponibile oggi che si è dimostrata attiva sul clone neoplastico riducendo in circa il 20-25% dei pazienti la carica allelica del JAK-2. Lo svantaggio è il costo e la modalità di somministrazione; oltre di questo, numerosi sono gli effetti collaterali che ne limitano l’impiego come la fatigue, la sindrome di tipo influenzale, la depressione che ne impongono la sospensione definitiva in oltre il 25% dei pazienti nel lungo termine.
La terza molecola è l’ANA; nata come farmaco antiaggregante negli anni 90 e dimostratasi attiva nel diminuire il numero delle piastrine circolanti. Tale molecola ha una attività molto buona agendo sulla liberazione delle piastrine da parte dei megacariociti midollari. In uno studio multicentrico si sarebbe dimostrata una inferiorità rispetto alla HU nel ridurre il numero degli episodi trombotici arteriosi15. Ma una rivalutazione avrebbe confermato una pari efficacia delle due molecole (HU e ANA)16. Non essendo un chemioterapico è un farmaco utile nei pazienti giovani o di seconda linea in combinazione o meno dopo l’impiego dell’HU. Lo svantaggio di tale molecola, che viene somministrato per via orale, è la possibile attività β-mimetica intrinseca, che può determinare cefalea e sensazione di tachicardia: ciò impone una valutazione cardiologica prima dell’inizio di tale terapia.
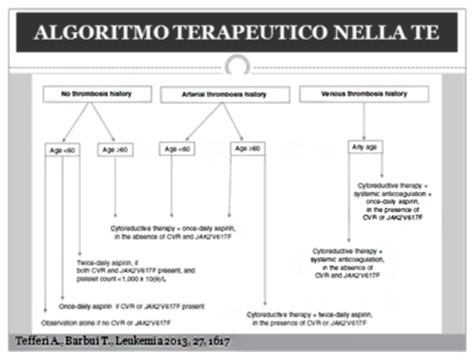
Si allega, a conclusione, l’algoritmo terapeutico proposto da Tefferi nel 2013 sulla terapia della TE17.
BIBLIOGRAFIA:
1. Klampfl T., Gisslinger H., Harutyunian AS et al.: Somatics mutations of calreticulin in myeloproliferative neoplasms. N.Engl.J.Med. 2013; 369: 2379-90.
2. Nangalia J., Massie CE, Baxter EJ et al.: Somatic CALR mutations in myeloproliferative Neoplasms with non-mutated JAK2. N.Engl.J.Med 2013; 369: 2391 – 2405
3. Rumi E., Pietra D., Ferretti V. et al.: JAK2 or CALRmutation status defines subtypes of essential thombocythemia with substantially different clinical course and outcome. Blodd 2014; dec 26 Epubahead of prints.
4. Thiele J., Kvasnicka H.M., Mullauer L. et al.: Essential thrombocitemia versus early myelofibrosis: a multicenter study to validate the WHO classification. Blood 117, 5710-16, 2011
5. Barbui T., Thiele J., Passamonti F. et al.: Survival and disease progression in Essential Thrombocitemia are significantly infuenced by accurate morphologic diagnosis: an International study. J. Cin. Oncol. 29: 3179-84, 2011.
6. Barbui T, Thiele J, Passamonti F et al.: Survival and disease progression in essential thrombocythemia are significantly influenced by accurate morphologic diagnosis: an international study.,. J Clin Oncol. 2011 Aug 10;29(23):3179-84.
7. Montanaro M., Latagliata R., Cedrone M. et al.: Thrombosis and survival in essential thrombocythemia: A regional study of 1,144 patients. Am.Jour. Hematol.; 2014;
8. Vannucchi AM: Insights into the pathogenesis and management of thrombosis in polycythemia vera and essential thrombocythemia.. Intern.Emerg.Med.; 2010 Jun; 5(3): 177-84. Epub 2009 Sep 30. Review.
9. Barbui T., Carobbio A., Rambaldi A., Finazzi G. et al.: Perspectives on thrombosis in essential thrombocythemia and polycythemia vera: is leukocytosis a causative factor? Blood. 2009 July 23; 114(4): 759–763
10. Palandri F, Polverelli N, Catani L et al.: Impact of leukocytosis on thrombotic risk and survival in 532 patients with essential thrombocythemia: a retrospective study. Ann Hematol. 2011 Aug; 90(8): 933-8.
11. Girodon F, Dutrillaux F, Broséus J et al.: Leukocytosis is associated with poor survival but not with increased risk of thrombosis in essential thrombocythemia: a population-based study of 311 patients. Leukemia. 2010 Apr; 24(4): 900-3.
12. De Stefano V, Za T, Rossi E. et al.: Leukocytosis is a risk factor for recurrent arterial thrombosis in young patients with polycythemia vera and essential thrombocythemia. Am J Hematol. 2010 Feb; 85(2): 97-100.
13. Andriani A., Latagliata R., Cedrone M. et al.: Clinical fea tures of essential thombocythemia with spleen enlargement: the experience of Latial group for the study of SMPC, Ph -. EHA 2013, abstr. 3
14. Latagliata R., Rago A., Spadea A.. et al.: Decisional flow with a scoring system to start platelet-lowering treatment in patients with essential thrombocythemia: long-term results. Int.J.Hematol.; 2009 Nov; 90(4): 486-91.
15. Harrison CN1, Campbell PJ, Buck G et al.: Hydroxyurea compared with anagrelide in high-risk essential thrombocythemia. N Engl J Med. 2005 Jul 7;353(1):33-45.
16. Gisslinger H1, Gotic M, Holowiecki J, et al.: Anagrelide compared with hydroxyurea in WHO-classified essential thrombocythemia: the ANAHYDRET Study, a randomized controlled trial. Blood. 2013 March 7; 121(10): 1720–1728
Alessandro Andriani Responsabile U.O.D. Ematologia, ASL RMA